
[ad_1]
A combination of the environment an individual experiences and their genetic predisposition determines the majority of their risk for various diseases. Large national efforts such as the UK Biobank have created large, public resources to better understand the links between environment, genetics and disease. This has the potential to help people better understand how to stay healthy, clinicians to treat disease, and scientists to develop new medicines.
One of the challenges of this process is how we capture the vast amount of clinical measurements – the UK Biobank has many petabytes of images, metabolic tests and medical records covering 500,000 individuals. To make the best use of this data, we need to be able to represent the information presented as concise, informative labels about important diseases and traits, a process called phenotyping. This is where we can use the ability of ML models to pick out subtle complex patterns in large amounts of data.
We have previously demonstrated the ability to use ML models for large-scale phenotyping of retinal diseases. However, these models were trained using labels from clinician reasoning, and access to clinical-grade labels is a limiting factor due to the time and cost required to create them.
Chronic Obstructive Pulmonary Disease Inference by Deep Learning on Raw Spirograms, Identifies Novel Genetic Loci and Refines Risk Models, published in Genetics of nature, we are pleased to present a method that learns accurate ML models for genetic disease discovery, even when noisy and unreliable labels are used. We demonstrate the ability to train ML models capable of phenotyping directly from raw clinical measurement and untrusted medical record information. This reduced reliance on medical domain experts for labeling greatly expands the range of applications of our technique to a panoply of diseases and has the potential to improve their prevention, diagnosis, and treatment. We present this method with ML models that can better characterize lung function and chronic obstructive pulmonary disease (COPD). Furthermore, we demonstrate the utility of these models by demonstrating a better ability to identify genetic variants associated with COPD, improved understanding of the biology behind the disease, and successful prediction of COPD-related outcomes.
ML for a deeper understanding of the breath
For this demonstration, we focused on COPD, the third leading cause of death worldwide in 2019, in which airway inflammation and airflow obstruction can progressively reduce lung function. Lung function in COPD and other diseases is measured by recording the volume an individual exhales over time (a recording called a spirogram; see example below). Although there are guidelines (called GOLD) for determining COPD status by exhalation, they use only a few, specific data points in the curve and use fixed thresholds for these values. Much of the rich data from these spirograms is neglected in this analysis of lung function.
We reasoned that ML models trained to classify spirograms would be able to more fully exploit the rich data presented and use more accurate and comprehensive measurements of lung function and disease than we have seen in other classification tasks such as mammography or histology. We trained ML models to detect whether an individual has COPD using complete spirograms.
 |
| Spirometry and COPD status review. Pulmonary function test spirogram showing forced expiratory volume-time spirogram (left), forced expiratory flow time spirogram (the middle) and interpolated forced expiratory flow volume spirogram (right). The profile of individuals with COPD is different. |
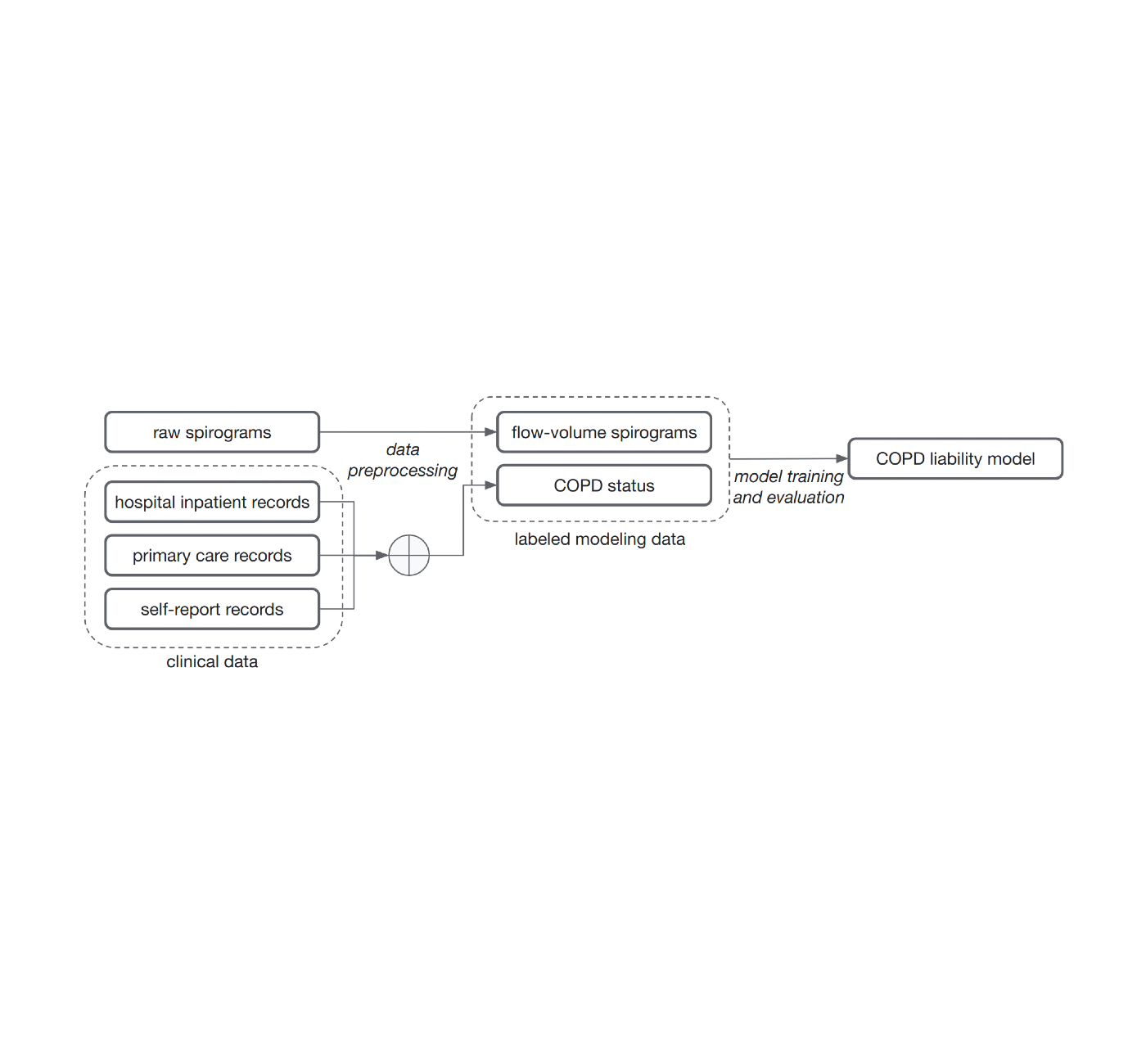
A common method for training models for this problem, supervised learning, requires associating patterns with labels. Determining these labels may require very time-constrained expert effort. For this work, to demonstrate that we do not necessarily need medically classified labels, we decided to use various widely available sources of medical record information to generate these labels without medical expert review. These labels are less reliable and noisy for two reasons. First, there are gaps in individuals’ medical records because they use multiple health services. Second, COPD is often underdiagnosed, meaning that many people with the disease would not be labeled as having it, even if we completed complete medical records. Nevertheless, we trained a model to predict these noisy labels from spirogram curves and model predictions as a quantitative COPD liability or risk score.
 |
| Noisy COPD status labels were derived using different medical record sources (clinical data). A COPD liability model is then trained to predict COPD status from the raw flow volume spirograms. |
Predicting outcomes in COPD
We then investigated whether the risk scores produced by our model could better predict binary COPD outcomes (for example, an individual’s COPD status, whether they were hospitalized for COPD or died from it). For comparison, we compared the model to expert-defined measurements needed to diagnose COPD, specifically FEV1/FVC, which compares specific points on the spirogram curve with a simple mathematical ratio. We observed an improvement in the ability to predict these outcomes, as shown in the precision-recall curves below.
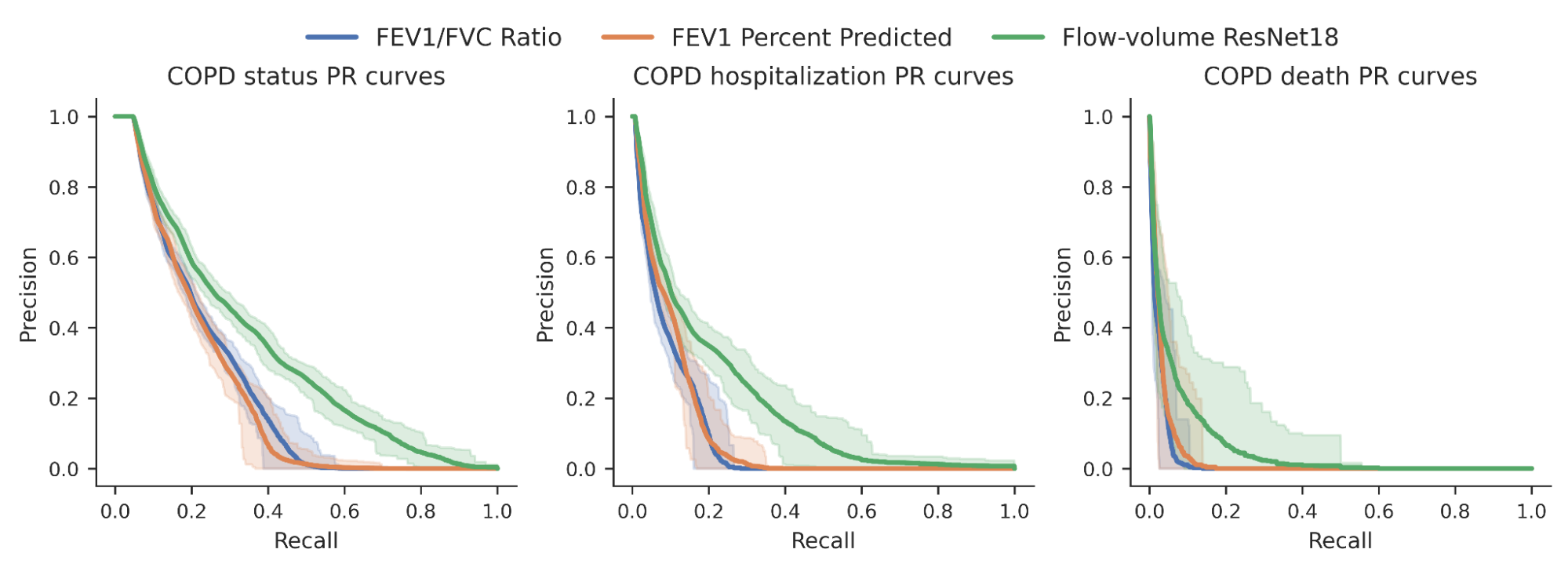 |
| Accurate recall curves for COPD status and outcomes for our ML model (green) compared with traditional measures. Confidence intervals are shown in light shading. |
We also observed that segregation of populations by COPD model score was predictive of all-cause mortality. This plot suggests that those at high risk of COPD die earlier from any cause, and the risk probably has implications beyond just COPD.
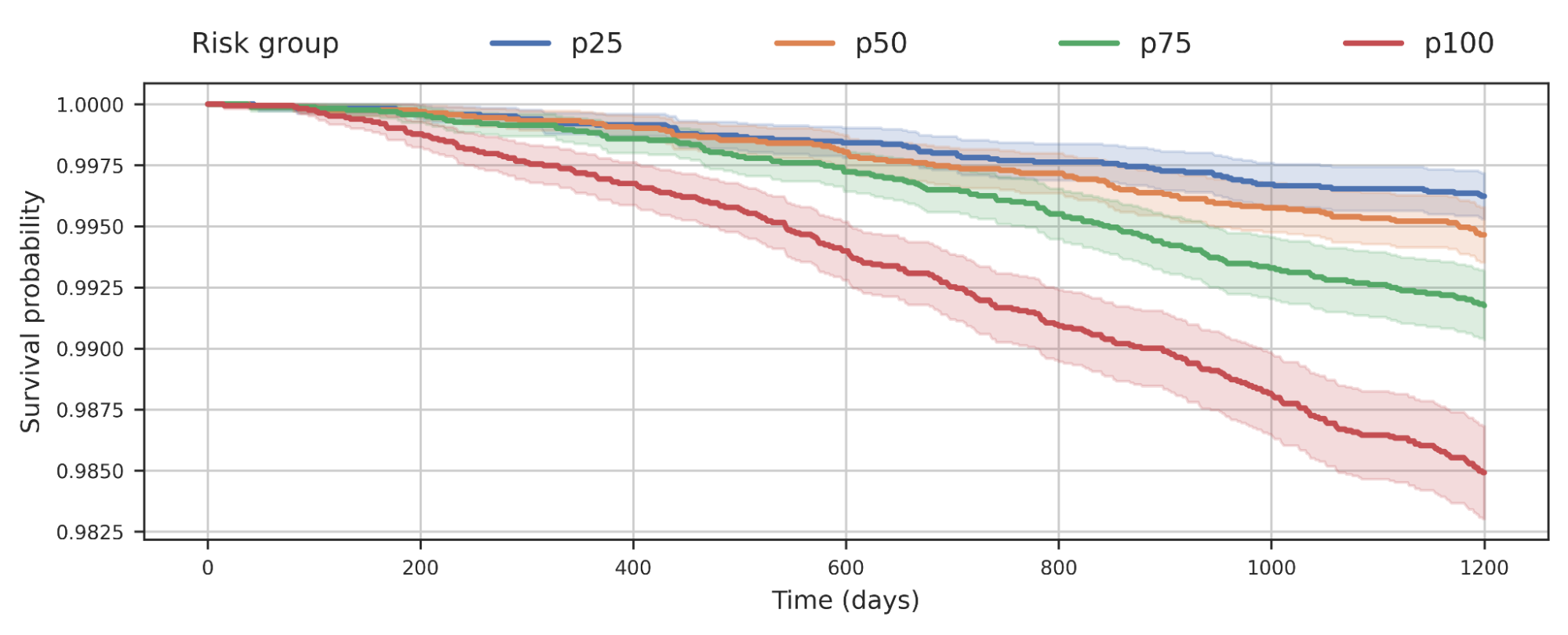 |
| Survival analysis of a cohort of UK Biobank individuals stratified by their COPD model predicted risk quartile. A decrease in the curve indicates that individuals in the group are dying over time. For example, p100 represents the 25% of the group with the greatest predicted risk, and p50 represents the 2nd quartile. |
Identifying genetic associations with COPD
As the goal of large-scale biobanks is to collect large amounts of phenotypic and genetic data, we also performed a test called a genome-wide association study (GWAS) to identify genetic associations with COPD and genetic susceptibility. GWAS measures the strength of the statistical association between a given genetic variant—a change in a specific DNA position—and observations (eg, COPD) in a group of cases and controls. Genetic associations discovered in this way can inform the development of drugs that alter gene activity or products, as well as expand our understanding of disease biology.
We have shown with our ML-phenotyping method that we not only rediscover almost all known COPD variants detected by manual phenotyping, but we also find many new genetic variants significantly associated with COPD. Additionally, we see good agreement on effect sizes for variants detected by both our ML approach and the manual (R2=0.93), which provides strong evidence for the validity of the newly found variants.
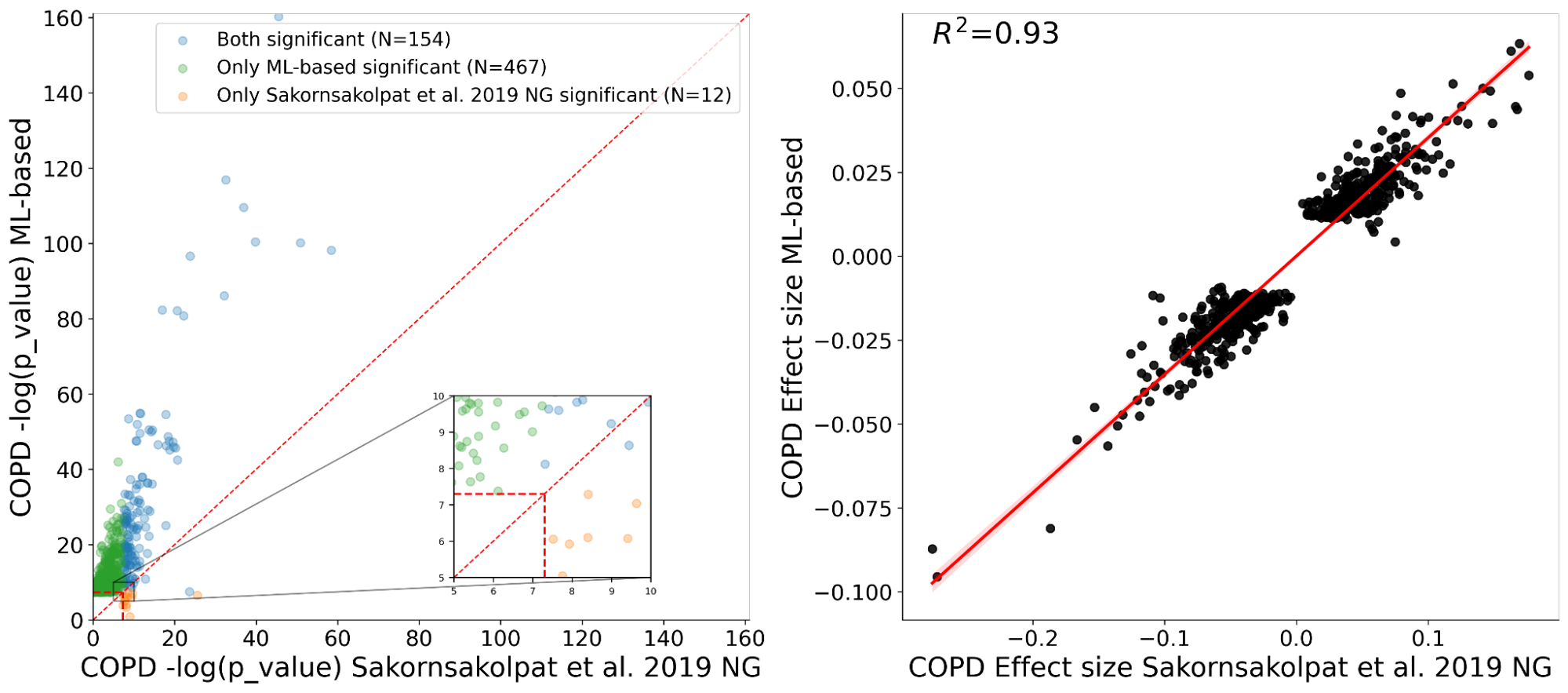 |
| left: Chart comparing statistical power of genetic discovery labels for our ML model (y-axis) with statistical power Guide labels from traditional research (x– axis). meaning above year = x The line indicates greater statistical power in our method. Green dots indicate important findings in our method that are not found using the traditional approach. Orange dots are important in the traditional approach, but not in ours. The blue dots are important in both. right: to estimate the association effect between our method (year-axis) and the traditional method (x-axis). Note that relative values are comparable between studies, but absolute numbers are not. |
Finally, our colleagues at Harvard Medical School and Brigham and Women’s Hospital further explored the plausibility of these findings on the possible biological role of new variants in the development and progression of COPD (more discussion of these insights can be found in Ref. ).
conclusion
We have shown that our earlier methods of phenotyping by ML can be extended to a wide range of diseases and can provide new and valuable insights. We made two key observations using this to predict COPD from spirograms and discovered new genetic insights. First, domain knowledge was not necessary to make predictions on raw medical data. Interestingly, we have shown that raw medical data is likely to be underutilized and the ML model can find patterns in it that are not captured by expert-defined measurements. Second, we don’t need medically classified labels; Instead, noise labels determined from widely available medical records can be used to generate clinically predictive and genetically informative risk scores. We hope that this work will greatly expand the field’s ability to use noise labels and improve our collective understanding of lung function and disease.
Acknowledgments
This work is the result of the collaboration of many contributors and institutions. We thank all participants: Justin Cosentino, Babak Alipanahi, Zachary R. McCaws, Corey I. McLean, Farhad Hormozdiar (Google), Davin Hill (Northeastern University), Tae-Hwi Schwantes-An, and Dongbing Lai (Indiana University), Brian D. Hobbs and Michael H. Cho (Brigham and Women’s Hospital and Harvard Medical School). We also thank Ted Yoon and Nick Furlot for reviewing the manuscript, Greg Corrado and Shravya Shetty for support, and Howard Young, Kavita Kulkarni, and Tammy Huynh for help with publishing logistics.
[ad_2]
Source link